Host factors
Severity of HIV disease after infection is variable. The majority of knowledge on the impact of host genetic variation on outcome of HIV-1 infection has been gained through studies of historical cohorts collected before the implementation of ART. An impact of host genetic variation on susceptibility to HIV-1 infection was identified early in the pandemic, with a major role attributed to the genes encoding HLA-B molecules and the chemokine receptor CCR5. Variants of genes encoding other chemokine receptors, as well as chemokines and cytokines, have been reported to influence HIV disease progression or transmission or HIV (Table 2). However, only variation within or near the HLA genes and, to a lesser extent, the CCR5 locus have been replicated in large genome-wide association studies.[100]
|
Table 2. Variants of genes encoding chemokines, chemokine receptors and cytokines associated with rate of HIV transmission or disease progression |
|||||
|
Gene variant |
Influence on natural history |
Putative effect |
|||
|
Site |
Codon |
Transmission |
Disease progression |
||
|
Chemokines |
|||||
|
SDF1 (CXCL12) |
3’UTR |
SDF1-3A’ |
Decrease |
Decrease |
? block CXCR4 |
|
RANTES (CCL5) |
Promoter |
G4 |
Increase |
Decrease |
? increase RANTES |
|
Chemokine receptor |
|||||
|
CCR5 |
Promoter |
59029 G |
Decrease |
Decreased promoter activity |
|
|
CCR5P1 |
Increase |
||||
|
ORF |
59356 T CCR5∆32 |
Increase MTCT |
Decrease |
||
|
CCR2 |
ORF |
CCR2-64I |
Decrease Delayed progression to AIDS then accelerated progression |
Linked with CCR5 promoter polymorphism, however mechanism undefined. |
|
|
Cytokines |
|||||
|
IL-4 |
Promoter |
IL-4 589 T |
Decrease, increased acquisition of X4 phenotype, associated with delayed progression to AIDS and then accelerated progression |
? increased IL-4 production with resultant decrease CCR5 and AIDS then increase CXCR4 expression |
|
|
IL-10 |
Promoter |
IL-10 5’A |
Increase |
Increase |
Decreased IL-10 inhibition of HIV |
|
IL: interleukin; MTCT: mother-to-child transmission; ORF: open reading frame; RANTES: regulated on activation, normal T cell expressed and secreted; SDF: stromal-derived factor; UTR: untranslated region. |
|||||
Chemokine receptors and chemokines associated with HIV-1 disease progression
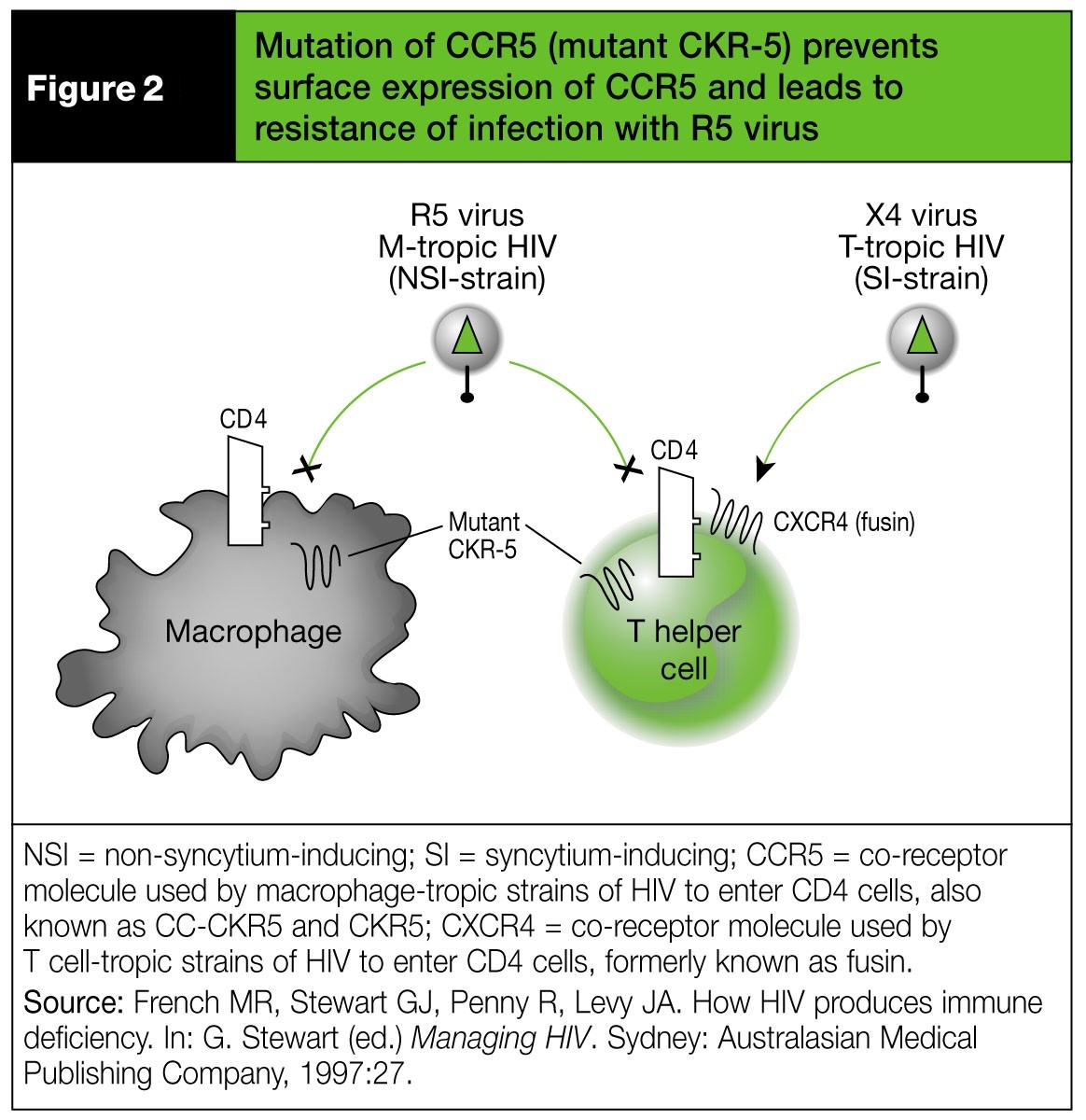
The first reported variant of the CCR5 gene was a 32-base-pair deletion (CCR5∆32). This gene variant encodes for a CCR5 molecule that is not expressed on the cell surface. The absence of CCR5 is the basis for the resistance of some individuals to infection with HIV-1 that uses CCR5 alone as a coreceptor (so called R5 viruses) (Figure 2).[101] However, homozygotes for CCR5∆32 can occasionally be infected with HIV isolates which use other coreceptors.[102] Cell-surface expression of CCR5 is reduced in heterozygotes (CCR5∆32/WT) and this is thought to be the basis of delayed progression to AIDS in these subjects. Variants may also occur in the promoter region of the CCR5 gene. Variants of the gene encoding the chemokine receptor CCR2 are also associated with delayed disease progression.[103] A chemokine that binds to CCR5, CCL3, also affects HIV acquisition and HIV disease progression. A lower CCL3L1 gene copy number is associated with higher susceptibility to HIV infection and disease progression.[104]
HLA-B alleles associated with HIV-1 disease progression
Several alleles of genes encoding HLA-B molecules have been shown to impact on the natural history of HIV-1 infection, particularly HLA-B*5701 and HLA-B*2705, which are associated with decreased rates of disease progression in caucasian people [105]. People with HLA-B*5701 are less likely to present with symptomatic acute HIV infection and are more likely to have broader and stronger T cell responses against HIV-1 peptides [106]. The association of HLA-B*5701 and other HLA-B alleles with rates of HIV disease progression reflects differences in the capacity of the encoded molecules to present HIV peptides to CD8+ T cells. [105]
Gender differences
Some studies have demonstrated that women develop AIDS at higher CD4+ T cell counts than men.[107] However, this difference may be explained by decreased access to care rather than biological effects. Conversely, other studies have demonstrated that, for a given CD4+ T cell count, women have up to a 0.3 log10 lower plasma HIV viral load than men.[108] This difference is most apparent in the 4 years following seroconversion.[109] After that time, women experience greater rises in plasma HIV viral load. This late rise in HIV viral load in women may account for the lack of gender differences in disease progression observed in other cross-sectional studies.[110] The underlying mechanisms for these observed differences remain undefined. No gender differences have been demonstrated in clinical progression following seroconversion.[111]
Viral factors
Variation in HIV influencing HIV disease progression
Multiple factors affecting the structure and function of HIV have been associated with the rate of HIV disease progression, including deletions in certain viral genes; coreceptor usage; viral subtype and replicative capacity. Slower disease progression rates were observed in people infected with HIV carrying deletions in the Nef gene (Nef-deleted viruses).[112] People infected with dual tropic (ie. mixed R5 and X4 tropic) viral populations progressed to AIDS 3.8 times more rapidly, independent of CD4+ T cell count and HIV viral load, than those with only R5-tropic viruses at baseline.[113] This is because viruses that use the CXCR4 coreceptor are more pathogenic than other strains: they form syncytia (giant, multinucleated cells) in vitro and, therefore, were previously described as syncytium-inducing variants. In contrast, R5 viruses are less pathogenic than X4 viruses: they do not form syncytia in vitro and, therefore, were previously referred to as non-syncytium-inducing strains. Transmission of X4 strains, which occurs rarely, is associated with accelerated disease progression. The viral coreceptor usage changes from R5 to X4 in up to 40% of patients during the course HIV disease and is associated with acceleration in CD4+ T cell loss and disease progression. [114] Subjects with syncytium-inducing (X4) variants are seven-times more likely to progress to AIDS over a 30-month period.
Some studies report that HIV-1 subtype is an independent predictor of HIV disease progression. Women with the Brazilian variant of subtype B HIV infection had a faster progression of HIV disease than women with other subtype B variants.[115] People with subtype A HIV infection were reported to have slower rates of disease progression relative to subtype D and other subtypes.[116] [117] [118] Viral subtypes may also differ in their capacity to be transmitted; for example, HIV-1 subtype C is associated with increased vaginal shedding.[119]
There is also some evidence that HIV replication capacity is associated with CD4+ T cell decline, though not with HIV viral load. People infected with HIV that had a high replicative capacity were more likely to have fast rates of CD4+ T cell decline, independent of plasma viral load.[120]
GB virus-C co-infection
The flavivirus GB virus-C (GBV-C), previously designated hepatitis G, may have a protective effect on HIV disease progression. No human disease has been associated with GBV-C infection. Epidemiological studies have described an association between GBV-C co-infection and decreased morbidity and mortality in individuals with HIV infection. [121] [122] [123] HIV-infected people with GBV-C co-infection have reduced mortality, increased survival post-AIDS and lower plasma HIV viral loads compared with HIV-infected people without GBV-C infection. Higher CD4+ T cell counts have been demonstrated in some [124] but not all [121] studies of people with HIV/GBV-C co-infection. As GBV-C and HIV share transmission routes, co-infection is common and has been demonstrated in between 16- 40% of people with HIV infection in the USA. This compares with a prevalence of GBV-C infection in 2% of volunteer blood donors and 20% of intravenous drug users in the USA.[125] [126]
Several mechanisms of interaction between GBV-C and HIV have been proposed, including altered cytokine profiles, HIV coreceptor expression, T-cell activation, Fas-mediated apoptosis and direct inhibition of HIV replication.[127] In vitro studies have demonstrated that GBV-C inhibits HIV replication by inducing the production of chemokines, which may inhibit HIV replication and reduce the expression of coreceptors on the surface of T-cell.[128] This is in keeping with the observation that persistent GBV-C virus infection is required for delayed progression, as people who clear GBV-C infection have higher mortality rates than those with persistent infection.[129] At present a causal relationship has not been proven.